
Radiation-Associated Sarcoma of Bone: A Report of Two Cases and Review of the Literature
Lauren K. Ehrlichman, MD, Caleb M. Yeung, BA, Erin F. Gillespie, MD, Mark C. Callanan, MD, John E. Ready, MD, Marco L. Ferrone, MD
Disclosure Information©2015 by The Orthopaedic Journal at Harvard Medical School
 PDF
PDFRadiation-associated sarcoma (RAS) is a rare tumor of mesenchymal tissue that arises in the setting of previous radiation exposure, most commonly for the treatment of another primary oncologic process. Although a rare complication of ionizing radiation, RAS has been increasingly recognized as cancer survival rates improve and cancer patients live longer following treatment. Patients will benefit from an improved understanding of the pathogenesis, natural history, and treatment of this condition. The cases described illustrate two examples of patients who developed RAS, one in the setting of previous external beam radiation and the other following proton beam radiation. Data regarding epidemiology, pathogenesis, natural history, and treatment of this condition are discussed.
Radiation-associated sarcoma (RAS) is a rare, malignant tumor of mesenchymal tissue that arises in the setting of radiation exposure, most commonly from radiation therapy (RT) for the treatment of another primary oncologic process. Representing 3-6% of all sarcomas, RAS can arise in any location and typically follows RT for lymphoma, breast cancer, or gynecologic cancer.1–9 RAS was one of the first solid cancers to be linked to ionizing radiation exposure, with the first cases described in the 1920s in patients who had received radiation therapy for benign bone conditions and in watch dial painters using radium paint.10,11 The association between radiation and malignancies was observed in the population that survived the World War II bombings. Radiation was identified as a precursor to malignancy in later studies of Japanese survivors of the atomic bombs, survivors of the Chernobyl nuclear accident, following occupational exposure to radiation, and after the use of radiation in medicine.12–14
The diagnostic criteria for RAS were first proposed in 1948 by Cahan in a case series of 11 sarcomas that manifested in irradiated bone.15 This definition of radiation-associated sarcoma was later revised in 1971 by Arlen et al. and in 1999 by Murray et al. Criteria for RAS include the following: 1) the sarcoma must have arisen in the setting of previous radiation exposure within the 5% isodose line of the irradiated field (i.e. the volume of tissue receiving at least 5% of the prescription dose), 2) the histologic features of the sarcoma must be of a different pathology than that of the primary malignancy, and 3) there must have been no evidence that the sarcoma was likely to have been present prior to the onset of radiation.10, 16, 17 The required length for latency period following initial treatment is controversial, as recent literature has suggested that as few as 6 months of latency can suffice for the diagnosis of radiation-associated sarcoma while other authors have suggested a 5-year latency period.15, 18
The absolute risk of RAS is low, with RAS thought to arise in approximately 0.1% of adults exposed to RT.19–21 However, there are several factors that can increase the risk of developing RAS. The incidence of RAS in children is significantly higher than in adults, which is likely due to longer survival after RT exposure as well as inherent biological differences including genetic predispositions that may have contributed to their first cancer.22–29 Among RAS arising from treatment of pediatric malignancies, osteosarcomas are the most common subtype and occur in the first 20 years following RT in children.22, 30 Genetic factors may also predispose to the subsequent development of RAS.23, 31–33 Other factors that may increase the risk of RAS include anthracycline or alkylating chemotherapy agents as well as exposure to industrial chemicals such as vinyl chloride, arsenic, and phenoxy herbicides.29, 34–37
Within all radiation-associated sarcomas, bone sarcomas represent approximately 20-30% of all RAS.2, 38–41 These are typically high-grade osteosarcomas, though fibrosarcomas and chondrosarcomas have also been reported with much less frequency.22, 42, 43 Other case studies have also shown RAS manifesting as extraosseous occurrences of osteosarcoma.44 The reported latency period between exposure and diagnosis of radiation-associated osteosarcoma is typically between 3-30 years.9, 45–48 A dose-related incidence of radiation-induced bone sarcomas with doses as low as 10 Gy has been shown, with strong associations described in the literature for doses of 30 Gy or greater.9, 30, 49–51 Radiation-associated sarcomas have been described in most bones in the body including the long bones of the upper and lower extremities as well as the spine, pelvis, hand, patella, sternum, clavicle, head and neck.48, 52–62
Radiation-associated osteosarcomas have been shown to arise secondary to radiotherapy treatments for various histologies, including the treatment of childhood leukemias, primary osteosarcoma, Ewing’s sarcoma, synovial sarcoma, and various carcinomas, including breast, uterine, bladder, nasopharyngeal, thyroid.45, 47, 57–59, 61–65 There is also data for secondary sarcoma after radiation therapy for benign disease, including heterotopic ossification which requires doses as low as 7 Gy in a single fraction.66 Recognition of secondary malignancy risk, particularly in younger patients with longer life expectancies, is one reason therapeutic radiation has evolved to treating primarily malignant processes in the past few decades.47, 50, 51
The last decade has seen a rapid increase in the use of intensity-modulated radiation therapy (IMRT), which involves multiple beam angles to enable highly conformal delivery of RT, or a treatment volume that follows the contour of the target. While this decreases the volume of tissue receiving higher radiation doses, it increases the volume of tissue receiving low doses, usually <5-10 Gy, because photons penetrate through the entire body. Interest in proton beam radiotherapy has increased in recent decades due to its unique physical property called the Bragg peak, whereby, as a heavy charged particle, the beam’s entire dose is deposited at the target while eliminating exit dose. Dosimetric studies have suggested that protons can achieve conformal treatment plans while minimizing the volume of tissue irradiated, thereby decreasing the predicted risk of second malignancy significantly.67 Proton therapy continues to be controversial particularly given its high cost and limited data regarding increased efficacy, though it has largely been accepted as superior in the treatment of most pediatric malignancies due to potential reduction in volume of tissue irradiated and theoretical decreased second malignancy risk. While we do not believe that proton therapy obviates the risk of RAS through any unique property other than reducing integral dose, to our knowledge there are still very few reports in the literature of cases of RAS occurring after proton therapy. This is despite decades of experience with proton therapy in centers such as Harvard’s Cyclotron at Massachusetts General Hospital and Loma Linda University Medical Center, where the field of proton therapy was pioneered.
The referenced cases illustrate examples of two young patients who underwent radiation therapy for malignancy and subsequently developed osteosarcoma, the first in the setting of previous external beam radiation and the second following proton beam therapy.
A 33-year-old woman with a history of acute myeloid leukemia (AML) diagnosed in August 2008 had been previously treated with induction chemotherapy with cytarabine and daunorubicin followed by allogeneic bone marrow transplant; for conditioning, she underwent total body irradiation in November 2008 with a total radiation dose of 1,375 cGy over 3.5 days as well as chemotherapy (cyclophosphamide, thiotepa, and antithymocyte globulin (ATG)). The donor marrow was reinfused following CD34 T-cell selection. She was noted to be doing well until mid-May 2014, at which time she developed rapidly worsening pain in her left thigh and groin. The pain progressed to the point of waking her at night and necessitating the use of a cane to ambulate.
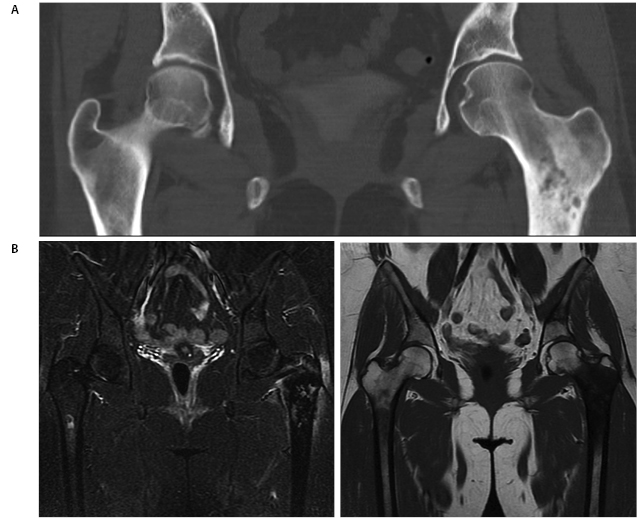
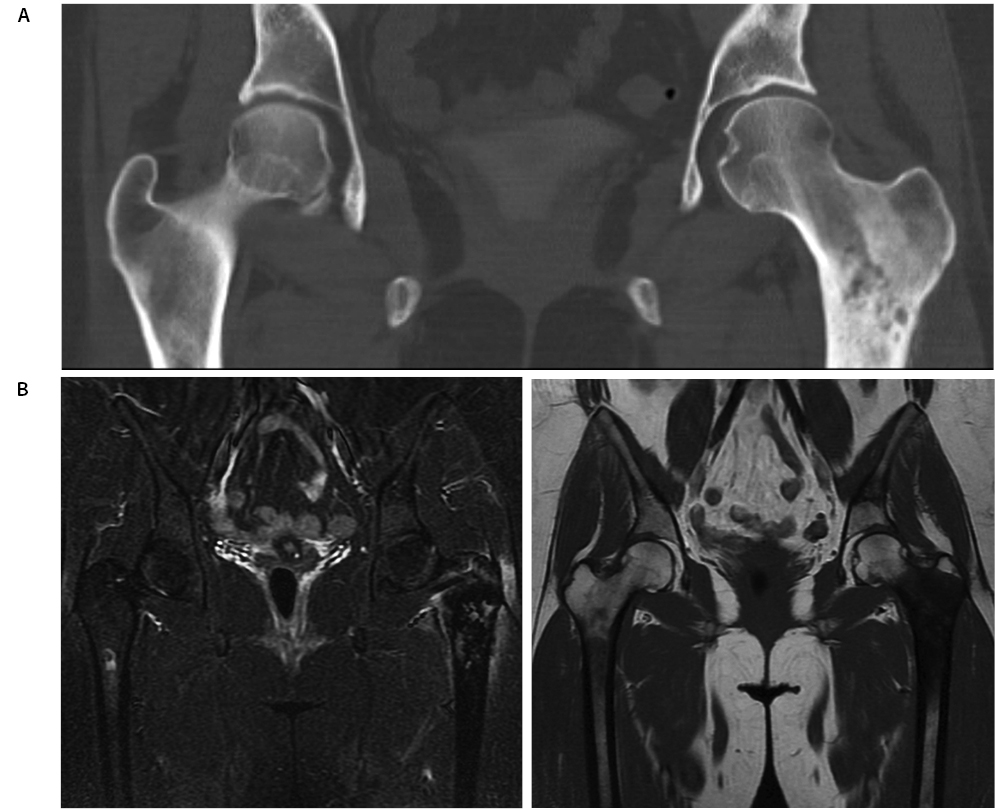
She presented to her primary care physician (PCP) in June 2014, at which time computed tomography (CT), magnetic resonance imaging (MRI), and a bone scan were obtained and demonstrated a lesion of the left proximal femoral metaphysis with extensive cortical destruction and a large enhancing extra-cortical soft tissue component (Figure 1). While multiple sclerotic lesions were visualized throughout the lower lumbar spine and pelvis, bone scan demonstrated uptake only in the left proximal femur.
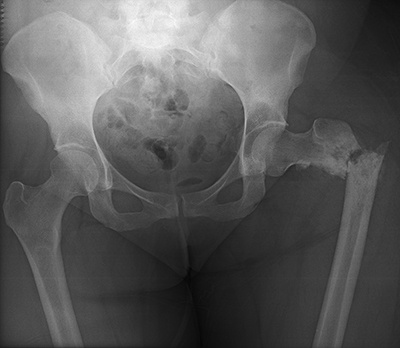
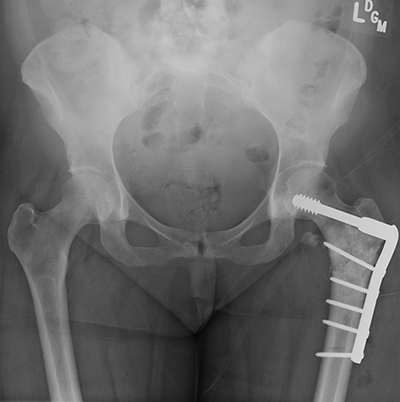
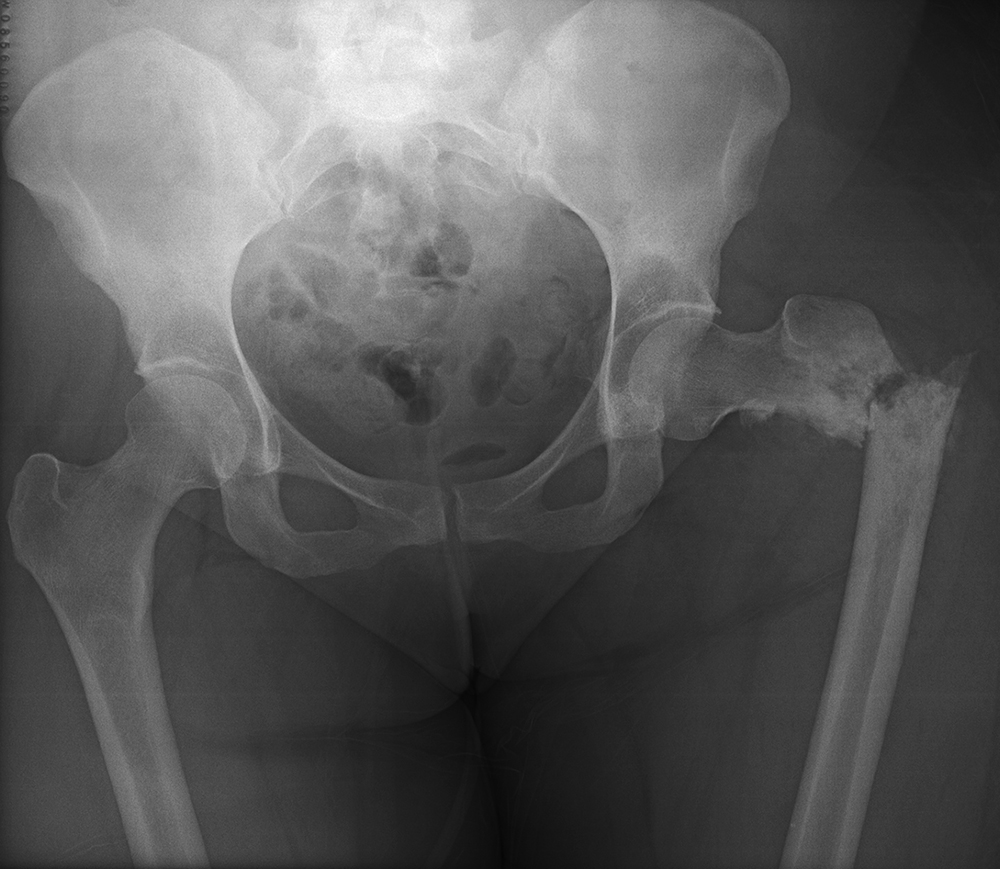
She returned to the cancer institute where she had previously been treated and was seen in consultation by the Orthopaedic Oncology service. During positioning to undergo CT-guided biopsy of the left proximal femur, she sustained a pathologic fracture through the lesion (Figure 2). She was admitted and underwent CT-guided needle biopsy of the soft tissue mass. Fine needle aspiration (FNA) was positive for malignant cells; immunohistochemistry was positive for Special AT-rich sequence-binding protein 2 (SATB2). Pathology demonstrated a poorly differentiated malignancy with immunohistochemistry consistent with osteosarcoma. The surgical team discussed with the family that operative stabilization of the fracture would allow the patient to undergo neoadjuvant chemotherapy prior to eventual proximal femoral resection. She subsequently underwent operative stabilization of the fracture with a 6-hole dynamic condylar screw system (Figure 3) and then began neoadjuvant chemotherapy with cisplatin and adriamycin 3 weeks after stabilization surgery. The first cycle of chemotherapy was complicated by profound thrombocytopenia requiring inpatient antibiotics and Neulasta (pegfilgrastim).
In October 2014, she returned to the operating room (OR) to undergo radical excision of the left proximal femur osteosarcoma, with reconstruction using a Stryker (Kalamazoo, Michigan) modular proximal femoral replacement cementless stem and a Stryker Tritanium cluster acetabular shell (Figure 4). Pathology review of the ~9.5cm osteosarcoma demonstrated 70% tumor necrosis. She resumed chemotherapy (C1D1 carboplatin/doxorubicin), with her last dose of doxorubicin in December 2014. She was then treated with ifosfamide. At her most recent follow-up in orthopaedic clinic 9 months following recurrence, she was able to ambulate with the use of one or two crutches.
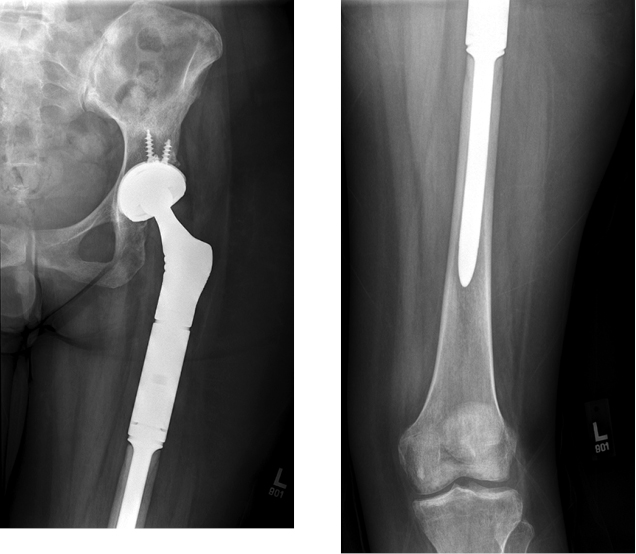
(A) Coronal, (B) Left posterolateral, (C) Right posterolateral
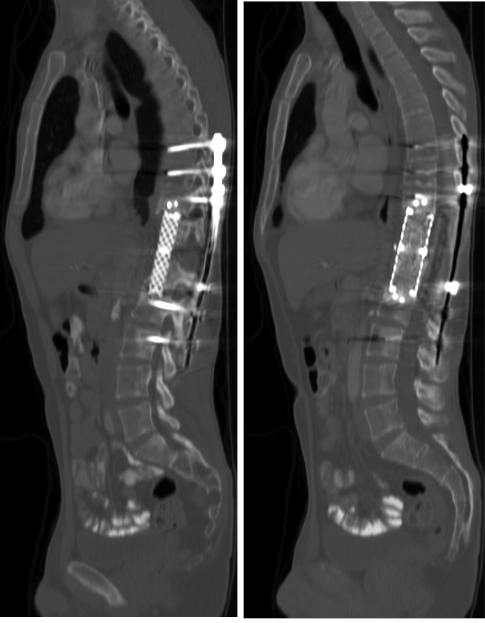
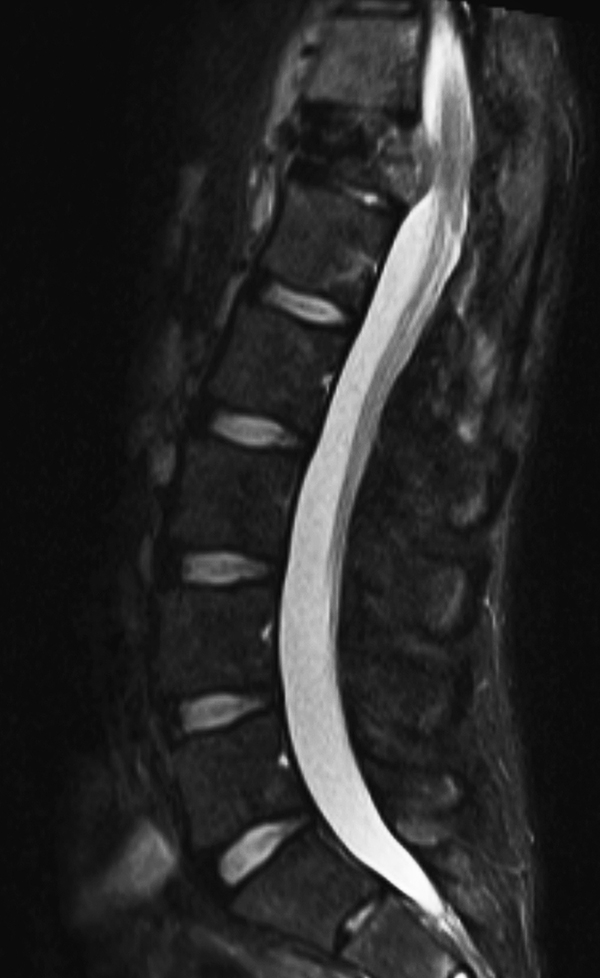
A 22-year-old man previously treated for L1 spinal ependymoma in 2006 with surgery and proton beam irradiation to 45 Gy presented with back pain 6 years after treatment. In February 2012 he developed back pain and was diagnosed with an atraumatic T12 compression fracture on CT and MRI with some epidural enhancement at the level of T12 (Figure 5). A CT-guided biopsy revealed necrotic bone with reactive marrow changes and focal new bone formation, consistent with osteoradionecrosis. There were no atypical cells or infiltrate suspicious for malignancy identified. In May 2012, he underwent kyphoplasty with no substantial improvement in his pain. Two weeks thereafter, he developed bilateral leg pain. In June 2012, he underwent transpedicular T12 decompression with placement of an expandable cage at T12 and T10-L2 posterior instrumentation. No gross tumor was observed.
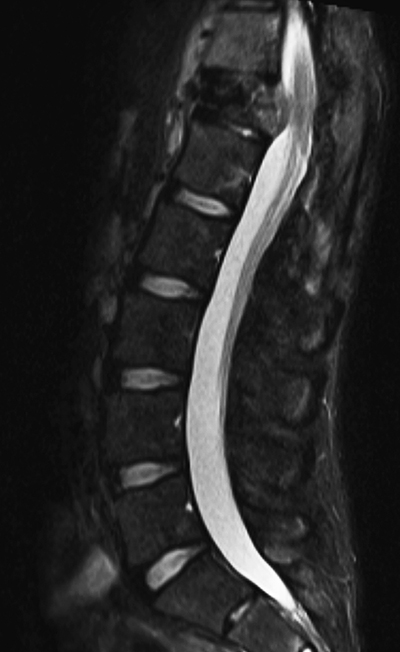
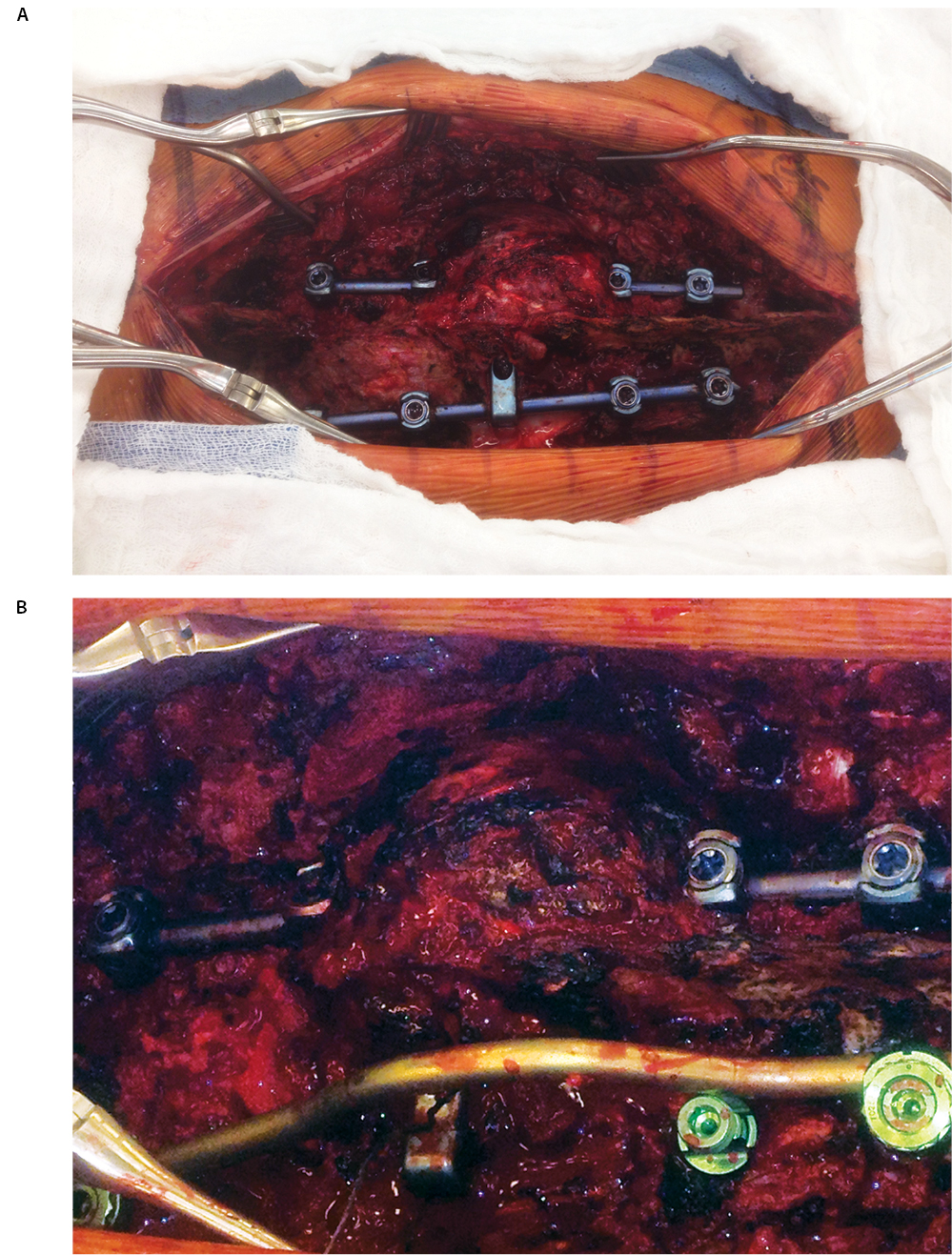
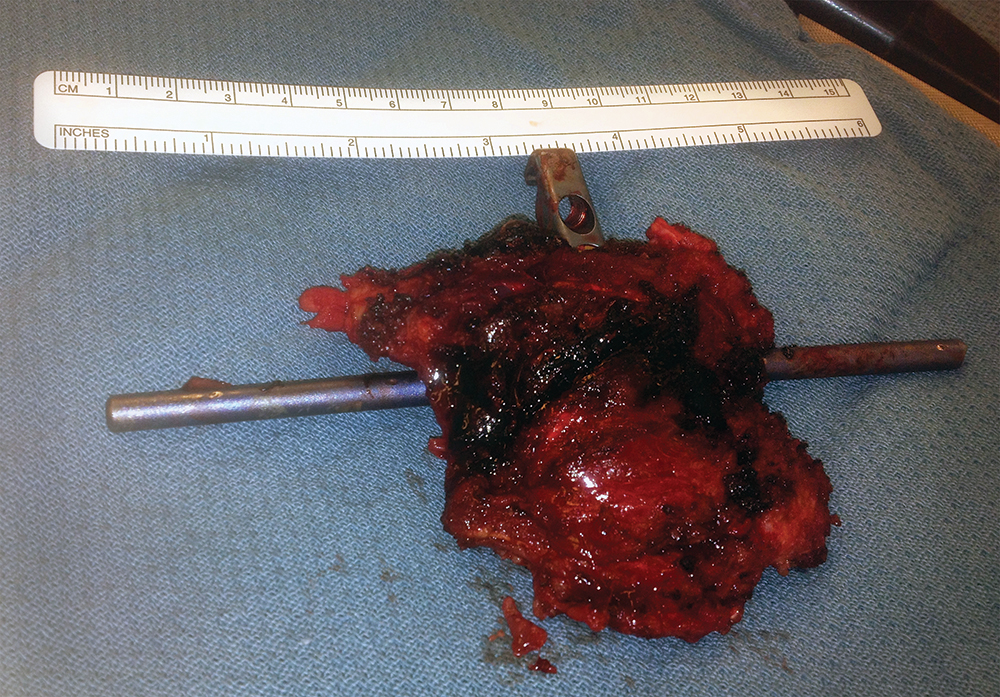
Post-operatively, he initially experienced a period of relief prior to the development of recurrent bilateral shooting pain and weakness; repeat MRI in October 2012 demonstrated a large enhancing mass involving T12 surrounding the cage, extending into T11 and L1, and surrounding the spinal canal, cord, and posterior hardware with anterior displacement of the great vessels (Figure 6). CT-guided biopsy was performed in October 2012, with pathology indicative of osteosarcoma. Shortly thereafter, he presented to our institution with difficulty initiating the flow of urine in addition to bilateral lower extremity weakness and pain with preserved ability to ambulate, low back pain, and weight loss. He was taken to the OR to undergo gross total resection of the tumor and decompression of the spinal cord, with removal of T10-L2 hardware followed by reinsertion and extension of the fusion and instrumentation to T8-L3, anterior instrumentation and fusion T11-L1, partial rib resections and rhizotomies, and partial corpectomies of T11 and L1 with full corpectomy of T12 (Figures 7 and 8).
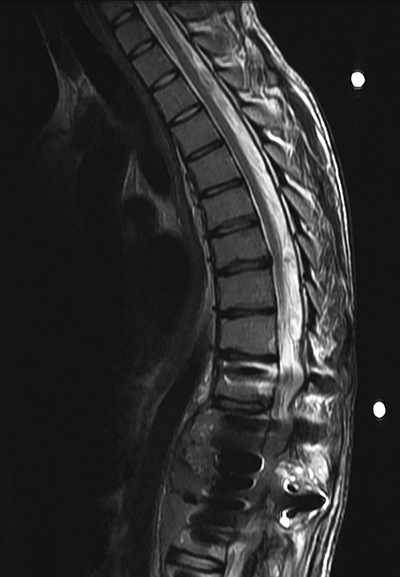
Demonstrates a large enhancing mass involving T12 surrounding the cage, extending into T11 and L1, and surrounding the spinal canal, cord, and posterior spinal hardware.
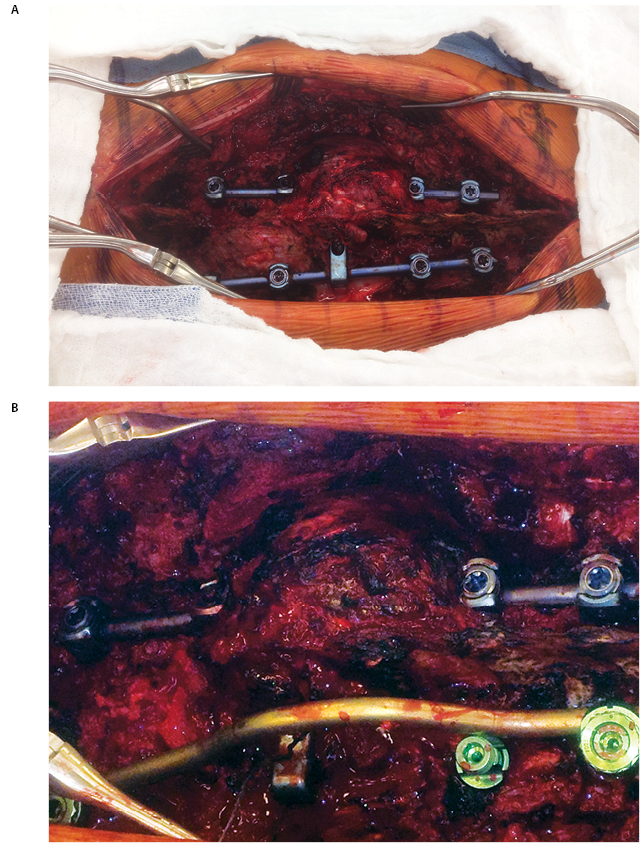
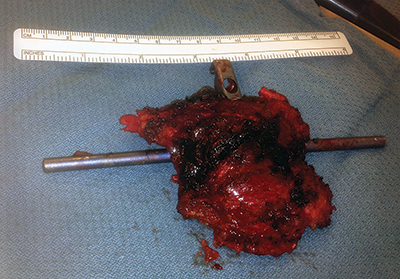
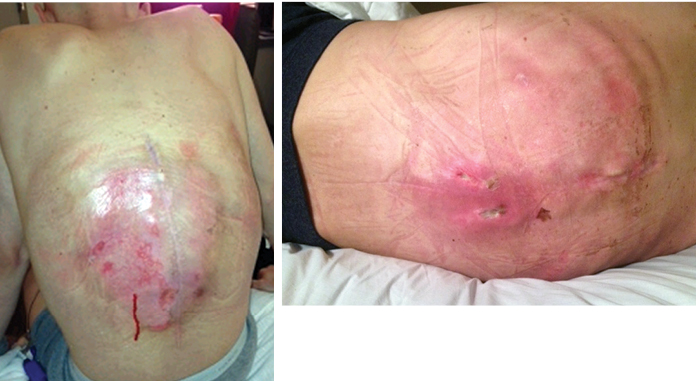
One month post-operatively, he began chemotherapy with adriamycin and cisplatin; four months post-operatively, he began proton beam radiation therapy to a prescription dose of 66.6 Gy, though tumor adjacent to the cord was limited due to spinal cord tolerance of 50 Gy. During the end stages of his treatment, he suffered failure of fixation with the cage telescoping and fracture of L1 and T11. Subsequently, the posterior rods fatigued. As a result of radiation therapy, he suffered from esophagitis, atrophy of his posterior musculature and radiation dermatitis (Figure 9). He returned to the OR in May 2013 to undergo stage one of a combined anterior-posterior approach, with day one consisting of removal of posterior spinal instrumentation at T11 and L1 as well as the posterior rods, followed by reinsertion of the rods and posterior spinal fusion from T8-L3 along with reduction of kyphosis from the pathologic fracture. Closure was facilitated by bilateral latissimus dorsi myocutaneous flaps and bilateral paraspinus muscle flaps. The following day, he returned to the OR for excision of the L1 mass, vertebrectomies of L1 and T11, removal and replacement of previous anterior spinal instrumentation with a vertebral interbody cage spanning T10-L2 and plating from T10-L12 (Figure 10).
Status post excision of the L1 mass, vertebrectomies of L1 and T11, removal and replacement of previous anterior spinal instrumentation with vertebral interbody cage spanning (A) T10-L2 and (B) plating from T10-L12
Advances in the treatment of malignancy have led to increasing numbers of long-term cancer survivors. As radiation therapy remains one of the mainstays of treatment for a variety of malignancies, the increase in the number of long-term survivors has led to an increase in the number of cases of RAS, leading to its apt name as an “iatrogenic disease of success.”10,11,46
RAS is an exceedingly rare disease with a poor prognosis. The overall incidence of RAS in patients who survive longer than five years after undergoing radiation therapy is approximately 0.1%.68 These tumors tend to be high grade and deep, limiting 5-year survival rates to 8.7% to 22%.20,69,70 While there is ample data describing RAS following external beam radiation with photons or X-rays, as in the first case described, the second case illustrates an example of a patient diagnosed with osteosarcoma following proton beam therapy. Rapid dose fall off at the proton Bragg peak gives proton therapy certain advantages over photon therapy (both conventional and IMRT). The proton beam enables dose escalation to tumor adjacent to critical structures, which has been utilized successfully in radiotherapy for base of skull tumors such as chordomas.71 Moreover, the “integral dose,” or total energy deposited in a patient from radiation therapy, is decreased significantly compared to photon therapy.72,73 In a study comparing radiation-associated secondary cancer risk resulting from IMRT and proton therapy in patients with either prostate or head-and-neck cancer, the estimated secondary cancer risk using passive scattering proton therapy was found to be significantly lower than that observed during IMRT treatment.74 Pediatric literature estimates a significant reduction in secondary malignancy risk with proton therapy.75 One Surveillance, Epidemiology, and End Results (SEER) study of patients treated at the Harvard Cyclotron between 1973-2001 reported 29 patients with a second malignancy after median follow-up of 6 years.76 While this number is expected to increase with longer follow-up, there is a paucity of reports of RAS in patients treated with proton beam therapy, thus making this second case particularly noteworthy.
The prognosis for radiation-associated sarcomas of the bone is widely reported to be poor. Bjerkehagen et al. found that factors contributing to poor survival in RAS include central tumor site, incomplete surgical resection, microscopic tumor necrosis, and the presence of metastases at the time of presentation.40,77 Some authors have shown that outcomes can be similar to those of similar patients with conventional osteosarcoma, particularly when treated with neoadjuvant chemotherapy.78,79 One report from the Rizzoli Institute involving 20 patients with radiation-associated osteosarcoma demonstrated a poorer prognosis for patients with radiation-associated osteosarcoma when compared to patients with sporadic osteosarcoma, with an 18% 3-year event-free survival in the sporadic group versus 0% in the radiation-associated osteosarcoma group. In their series of 27 patients, however, Lewis et al. did not find that prognosis correlated to chemotherapy response.80 These authors also found that better prognosis was seen with longer latency periods between radiation exposure and diagnosis.
Outcomes have also been shown to be related to treatment method, with a combination of surgical resection and intensive chemotherapy demonstrating improved results.40,78,81,82 The role of utilizing additional radiation therapy in the treatment of RAS is unclear, but in the second case reported here was used in conjunction with chemotherapy following surgical resection, the combination of which led to disease-free survival at 2.5 years of follow-up. In a series of 25 patients with RAS following treatment for nasopharyngeal carcinoma, all patients with positive margins following surgical resection (14/20 patients) underwent chemoradiation with 6 also undergoing brachytherapy; of these patients, 71.4% developed local recurrence.38 Thus, the optimal treatment algorithm for RAS in addition to surgical resection remains controversial, though may actually include radiotherapy.
Recent advances in radiotherapy have shown potential as future treatment methods for RAS. Of note, a recently published case study in Japan demonstrated effective treatment of a RAS with boron neutron capture therapy (BNCT).83 While a standard protocol has not been established for these rare tumors, other studies involving particle radiotherapy have also demonstrated promise in their therapeutic effects on osteosarcomas. It is thus conceivable that these therapies might also be leveraged in the future for treatment of RAS, particularly those which are unresectable.84,85 For now, however, early detection of RAS appears to offer the best hope for improved disease-free survival as there are currently no known preventive measures for RAS.86 As such, clinicians should have high suspicion and pursue aggressive workup when patients complain of any symptoms such as swelling or pain in an irradiated location.86
Although sarcomas satisfying the aforementioned 1948 Cahan criteria are likely to be radiation-associated, a gold-standard definition for RAS has yet to be developed. As such, research efforts have focused on whether radiation-associated sarcomas might be differentiated from sporadic sarcomas using differences in its molecular or genetic expression profile. Genetic sequencing of sporadic sarcomas versus radiation-associated sarcomas have demonstrated a high rate of TP53 mutations in RAS compared to sporadic sarcomas; furthermore, one study identified 135 genes via microarray analysis that could be used as a genetic signature to distinguish RAS from sporadic sarcoma with a 96% sensitivity and a 62% specificity.58,87,88 Many of these identified genes had roles in mitochondrial, detoxification, or antioxidant pathways, suggesting that chronic oxidative stress might be a defining characteristic of RAS.88
Another aspect of recent research efforts has involved the investigation of how benign post-radiation changes might be distinguished from malignant RAS tumors. A recent study using fluorescent in situ hybridization (FISH) to analyze angiosarcomas and atypical vascular lesions in patients who had received radiation for breast cancer revealed amplification of MYC, suggesting that this might be leveraged as a diagnostic tool for RAS in patients after radiation therapy.89 Other changes resulting from the direct effects of ionizing radiation might also be used for earlier detection and treatment of RAS. Indeed, one study demonstrated alterations in p53 in a murine model of RAS; other studies in mice have suggested the importance of RB1-mediated secretion of IL-6 and natural killer T (NKT) cell response in preventing development of RAS.90–92 Karyotype analysis of RAS has also implicated rearrangements of chromosome 3p in RAS.93 Future studies may investigate genetic alterations that occur in patients who develop RAS, as well as other potential contributing factors such as specific chemotherapy agents or genetic predisposition.
Although a rare complication of ionizing radiation, radiation-associated sarcoma has been increasingly recognized as radiation therapy becomes more common and cancer survival improves. Patients will benefit from an improved understanding of the pathogenesis, natural history, and treatment of this condition.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}